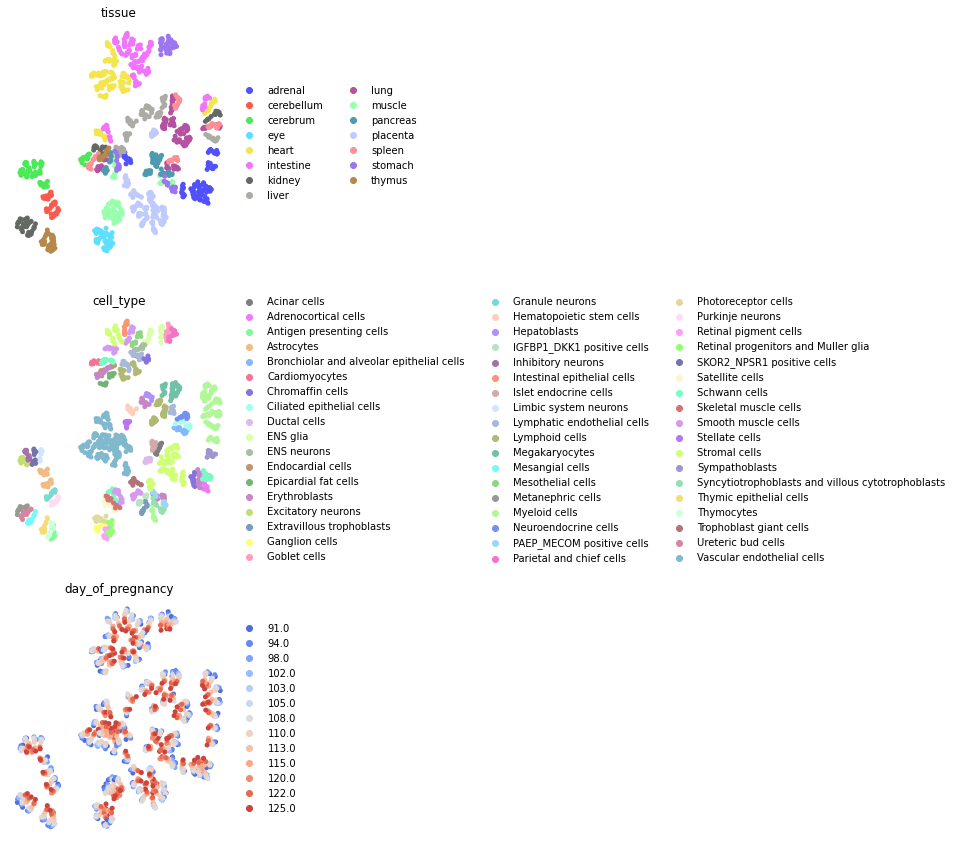Using biolord for -omics data#
This code presents an application of biolord to chromatin accessibility single-cell data.
We train a Biolord model on a fetal chromatin accessibility single-cell atlas [DHD+20] to obtain a meaningful disentangled latent representation.
import warnings
warnings.simplefilter("ignore", UserWarning)
import os
import sys
import scanpy as sc
from muon import atac as ac
import anndata
import numpy as np
import pandas as pd
import re
import seaborn as sns
import matplotlib.pyplot as plt
import itertools
import scipy.spatial as sp, scipy.cluster.hierarchy as hc
import biolord
Setup the AnnData#
atac = sc.read(
"adata_atac.h5ad",
backup_url="https://figshare.com/articles/dataset/atac-tissue-age-celltype/23702631",
)
fig, axs = plt.subplots(3, 1, figsize=(4, 15))
for i, c in enumerate(["tissue", "cell_type", "day_of_pregnancy_cat"]):
sc.pl.umap(atac, color=[c], ax=axs[i], show=False)
axs[i].set_axis_off()
plt.tight_layout()
plt.show()
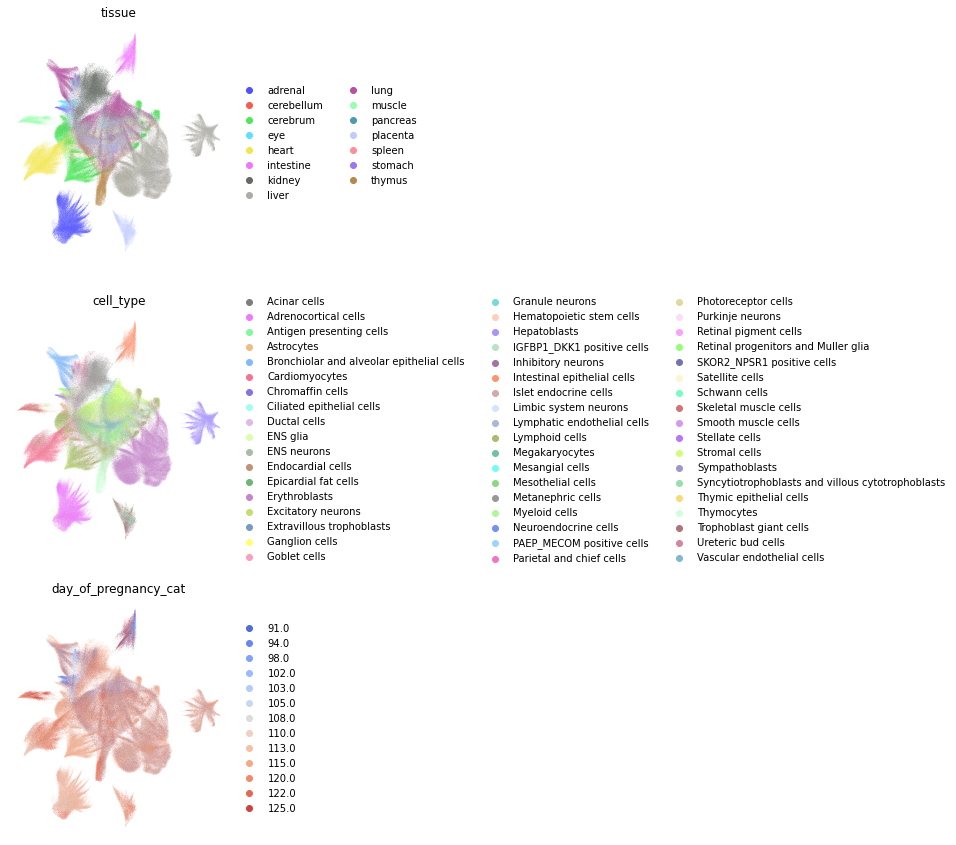
By calling biolord.Biolord.setup_anndata() we set the supervised attributes used for disentanglement.
The function takes as input:
adata: the adata object for the setting.ordered_attributes_keys: the keys inobsorobsmdefining ordered attributes.categorical_attributes_keys: the keys inobsdefining categorical attributes.layer: the keylayers(or simply “X” forX) we want to take measured features from.
biolord.Biolord.setup_anndata(
adata=atac,
ordered_attributes_keys=["day_of_pregnancy"],
categorical_attributes_keys=["tissue", "cell_type"],
layer="counts"
)
Run Biolord#
Instantiate a Biolord model#
We instantiate the model given the module_params.
These are parameters required to construct the model’s module, the various networks included in a Biolord model. Here we make sure to pass gene_likelihood=Poisson to model the peak counts[MFTG22].
module_params = {
"decoder_width": 512,
"decoder_depth": 6,
"attribute_nn_width": 256,
"attribute_nn_depth": 2,
"unknown_attribute_noise_param": 1e0,
"seed": 42,
"n_latent_attribute_ordered": 16,
"n_latent_attribute_categorical": 16,
"gene_likelihood": "poisson",
"reconstruction_penalty": 1e1,
"unknown_attribute_penalty": 1e0,
"attribute_dropout_rate": 0.1
}
model = biolord.Biolord(
adata=atac,
n_latent=128,
model_name="atac_poisson",
module_params=module_params,
split_key="split_random",
)
Train the model#
To train the model we provide trainer_params. These are paramters which dictate the training regime, e.g., learning rate, weight decay and scheduler type.
trainer_params = {
"n_epochs_warmup": 0,
"decoder_lr": 1e-4,
"decoder_wd": 1e-4,
"attribute_nn_lr": 1e-2,
"attribute_nn_wd": 4e-8,
"step_size_lr": 45,
"cosine_scheduler": True,
"scheduler_final_lr": 1e-5,
}
model.train(
max_epochs=100,
batch_size=1024,
plan_kwargs=trainer_params,
early_stopping=False,
enable_checkpointing=False,
early_stopping_patience=20,
check_val_every_n_epoch=10,
num_workers=1,
)
Epoch 100/100: 100%|██████████| 100/100 [1:16:15<00:00, 44.93s/it, v_num=1, generative_mean_accuracy=0, generative_var_accuracy=0, biolord_metric=0, reconstruction_loss=2.42e+3, unknown_attribute_penalty_loss=128, val_generative_mean_accuracy=0.812, val_generative_var_accuracy=0.125, val_biolord_metric=0.469, val_reconstruction_loss=2.44e+3, val_unknown_attribute_penalty_loss=6.07e-17]Epoch 100/100: 100%|██████████| 100/100 [1:16:15<00:00, 45.76s/it, v_num=1, generative_mean_accuracy=0, generative_var_accuracy=0, biolord_metric=0, reconstruction_loss=2.42e+3, unknown_attribute_penalty_loss=128, val_generative_mean_accuracy=0.812, val_generative_var_accuracy=0.125, val_biolord_metric=0.469, val_reconstruction_loss=2.44e+3, val_unknown_attribute_penalty_loss=6.07e-17]
Explore the latent space#
Obtain latent space representations#
Concatenate all possible combinations of cell_type, tissue and day_of_pregnancy.
We need to treat differently the categorical and ordered attributes:
categorical_attributes: We use
get_categorical_attribute_embeddings()which takes as inputattribute_keyand provides the latent vectors of all categories.ordered_attributes: We use
get_ordered_attribute_embedding()which requires along withattribute_keythevals, the desired values to get the vectors for.
attribute_keys_categorical = ["tissue", "cell_type"]
attribute_keys_continuous = ["day_of_pregnancy"]
attribute_keys = [*attribute_keys_categorical, *attribute_keys_continuous]
transf_embeddings_attributes = {}
for attribute_ in attribute_keys_categorical:
transf_embeddings_attributes[attribute_] = sc.pp.scale(model.get_categorical_attribute_embeddings(attribute_key=attribute_))
for attribute_ in attribute_keys_continuous:
transf_embeddings_attributes[attribute_] = sc.pp.scale(model.get_ordered_attribute_embedding(
attribute_key=attribute_,
vals=np.sort(atac.obs[attribute_].unique())[:, np.newaxis]
))
attribute_model_keys = {}
attribute_model_keys_maps = {}
for attribute_ in attribute_keys_categorical:
attribute_model_keys[attribute_] = list(model.categorical_attributes_map[attribute_].keys())
attribute_model_keys_maps[attribute_] = model.categorical_attributes_map[attribute_]
for attribute_ in attribute_keys_continuous:
cats = list(np.sort(atac.obs[attribute_].unique()))
attribute_model_keys[attribute_] = cats
attribute_model_keys_maps[attribute_] = {cat: i for i, cat in enumerate(cats)}
keys = list(itertools.product(*[attribute_model_keys[attribute_] for attribute_ in attribute_keys]))
transf_embeddings_attributes_dict = {
"_".join([str(k) for k in key_]): np.concatenate((
[
transf_embeddings_attributes[attribute_][attribute_model_keys_maps[attribute_][key_[ai]], :] for ai, attribute_ in enumerate(attribute_keys)
]
), 0)
for key_ in keys
}
transf_embeddings_attributes = [
np.concatenate((
[
transf_embeddings_attributes[attribute_][attribute_model_keys_maps[attribute_][key_[ai]], :] for ai, attribute_ in enumerate(attribute_keys)
]
), 0)
for key_ in keys
]
We create an AnnData object to analyze the embedding.
attr = np.asarray(transf_embeddings_attributes)
adata_emb = anndata.AnnData(X=attr, dtype=attr.dtype)
adata_emb.obs_names = ["_".join([str(k) for k in key]) for key in keys]
adata_emb.obs["tissue"] = [key[0] for key in keys]
adata_emb.obs["cell_type"] = [key[1] for key in keys]
adata_emb.obs["day_of_pregnancy"] = [key[2] for key in keys]
adata_emb
AnnData object with n_obs × n_vars = 10530 × 48
obs: 'tissue', 'cell_type', 'day_of_pregnancy'
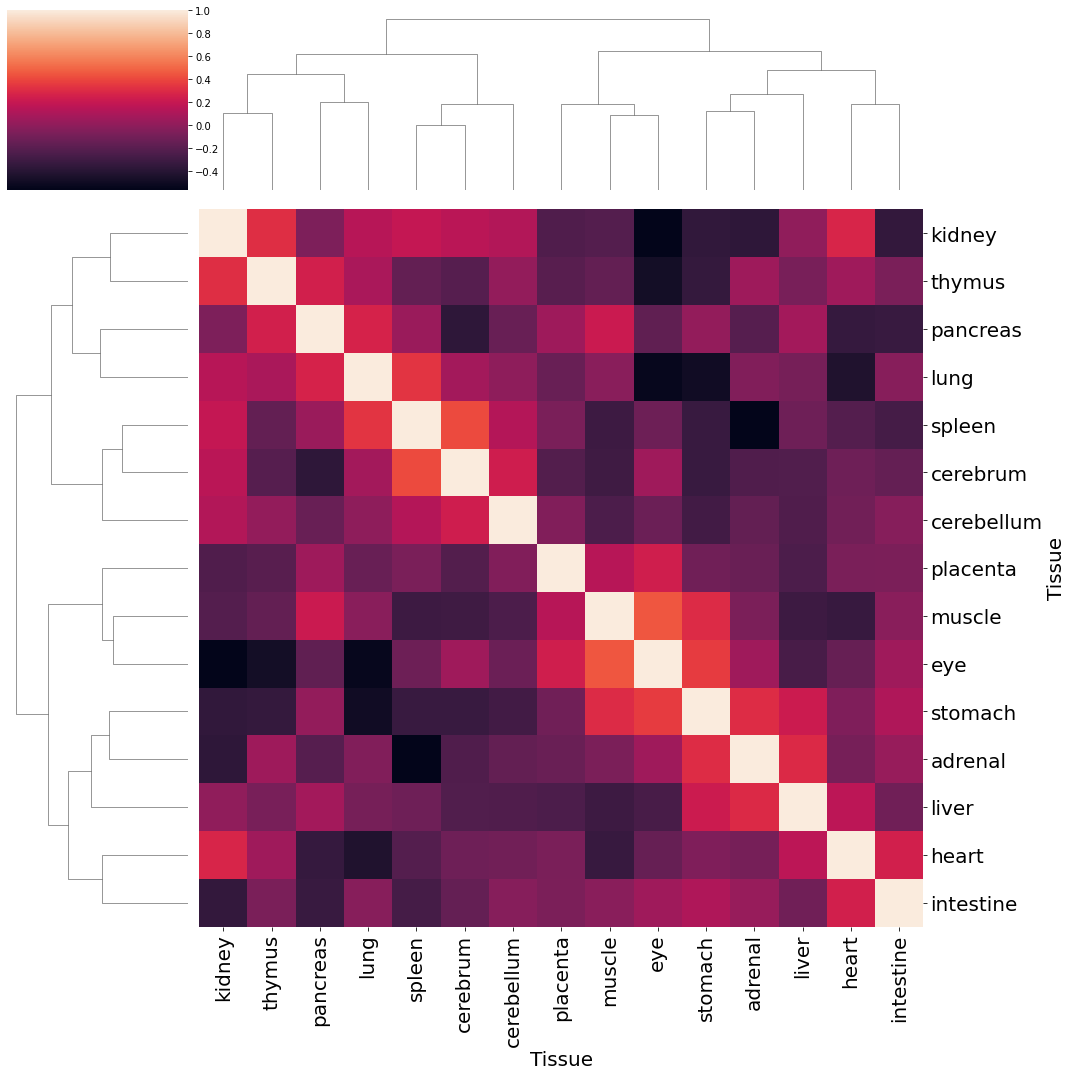
Visualize inner attribute relationships#
We first assess the correlations within each attribute
df = pd.DataFrame(attr, index=adata_emb.obs_names)
df = df.iloc[:, :16]
df["tissue"] = [key[0] for key in keys]
df_agg = df.groupby("tissue").mean().T
linkage = hc.linkage(df_agg.corr(), method='complete', optimal_ordering=True)
h = sns.clustermap(df_agg.corr(), row_linkage=linkage, col_linkage=linkage, yticklabels=1, xticklabels=1, figsize=(15, 15))
h.ax_heatmap.set_yticklabels(h.ax_heatmap.get_yticklabels(), rotation=0, fontsize=20)
h.ax_heatmap.set_xticklabels(h.ax_heatmap.get_xticklabels(), rotation=90, fontsize=20)
h.ax_heatmap.set_ylabel("Tissue", fontsize=20)
h.ax_heatmap.set_xlabel("Tissue", fontsize=20)
plt.tight_layout()
plt.show()
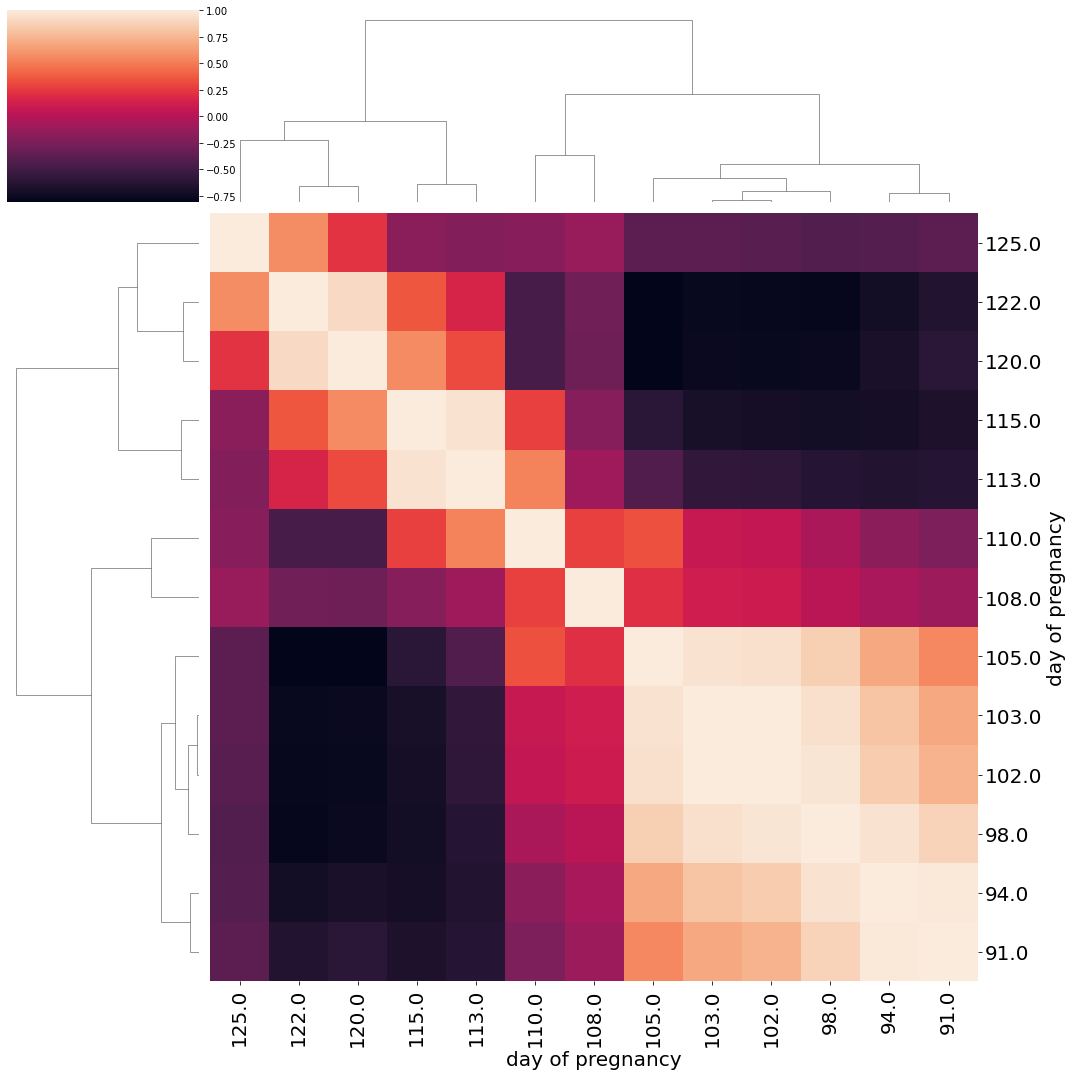
df = pd.DataFrame(attr, index=adata_emb.obs_names)
df = df.iloc[:, 32:]
df["day_of_pregnancy"] = [key[2] for key in keys]
df_agg = df.groupby("day_of_pregnancy").mean().T
linkage = hc.linkage(df_agg.corr(), metric="euclidean", method='complete', optimal_ordering=True)
h = sns.clustermap(df_agg.corr(), row_linkage=linkage, col_linkage=linkage, yticklabels=1, xticklabels=1, figsize=(15, 15))
h.ax_heatmap.set_yticklabels(h.ax_heatmap.get_yticklabels(), rotation=0, fontsize=20)
h.ax_heatmap.set_xticklabels(h.ax_heatmap.get_xticklabels(), rotation=90, fontsize=20)
h.ax_heatmap.set_ylabel("day of pregnancy", fontsize=20)
h.ax_heatmap.set_xlabel("day of pregnancy", fontsize=20)
plt.tight_layout()
plt.show()
Studying relations between attributes#
We turn to study the relationships between attributes by considering the concatenation of the latent vectors, such that each data point represent a combination of cell_type, tissue and day_of_pregnancy. Importantly, we restrict this to combinations of cell_type and tissue observed in the data.
adata_emb.obs["tissue"] = adata_emb.obs["tissue"].astype("category")
adata_emb.obs["cell_type"] = adata_emb.obs["cell_type"].astype("category")
adata_emb.obs["day_of_pregnancy"] = adata_emb.obs["day_of_pregnancy"].astype("category")
adata_emb.obs["day_of_pregnancy_cont"] = adata_emb.obs["day_of_pregnancy"].copy().astype(np.float16)
idx = []
for ti, tissue in enumerate(adata_emb.obs["tissue"].cat.categories):
cts = atac[atac.obs["tissue"].isin([tissue])].obs["cell_type"].unique()
idx.append(list(np.where((adata_emb.obs["tissue"].isin([tissue])) & (adata_emb.obs["cell_type"].isin(cts)))[0]))
adata_emb = adata_emb[np.concatenate(idx)]
sc.pp.pca(adata_emb)
sc.pp.neighbors(adata_emb, use_rep="X")
sc.tl.umap(adata_emb)
palletes = {
"tissue": [atac.uns["tissue_colors"][np.where(atac.obs["tissue"].cat.categories == cat)[0][0]] for cat in adata_emb.obs["tissue"].cat.categories],
"cell_type": [atac.uns["cell_type_colors"][np.where(atac.obs["cell_type"].cat.categories == cat)[0][0]] for cat in adata_emb.obs["cell_type"].cat.categories],
"day_of_pregnancy": [atac.uns["day_of_pregnancy_cat_colors"][np.where(atac.obs["day_of_pregnancy_cat"].cat.categories == cat)[0][0]] for cat in adata_emb.obs["day_of_pregnancy"].cat.categories],
}
fig, axs = plt.subplots(3, 1, figsize=(4, 15))
for i, c in enumerate(["tissue", "cell_type", "day_of_pregnancy"]):
sc.pl.umap(
adata_emb,
color=[c],
size=100,
palette=palletes[c],
ax=axs[i],
show=False
)
axs[i].set_axis_off()
plt.tight_layout()
plt.show()