Using biolord-classify pipeline#
Here we train a biolord-classify model on the spatio-temporally resolved single cell atlas of the Plasmodium liver stage [AZLBH+22] to extend the classification of Abortive state (provided for 36 hpi) to earlier stages of infection (24 and 30 hpi).
import pandas as pd
import numpy as np
import scanpy as sc
from scipy.stats import ttest_rel
import seaborn as sns
import matplotlib.pyplot as plt
import warnings
import biolord
from statannotations.Annotator import Annotator
warnings.simplefilter("ignore", UserWarning)
warnings.simplefilter("ignore", FutureWarning)
Setup the AnnData#
adata = sc.read("adata_abortive.h5ad", backup_url="https://figshare.com/ndownloader/files/39375752")
fig, axs = plt.subplots(1, 4, figsize=(20, 4))
for i, c in enumerate(["coarse_time", "zone", "abortive_state", "stress_score"]):
sc.pl.umap(adata, color=[c], ax=axs[i], show=False)
axs[i].set_axis_off()
plt.tight_layout()
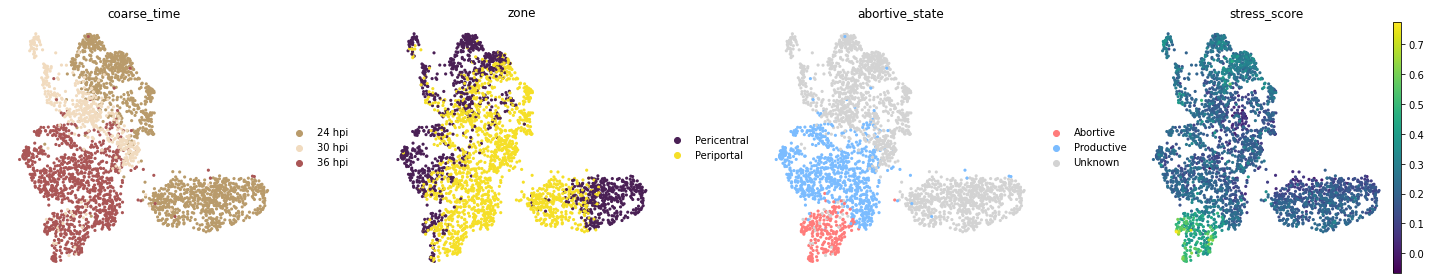
As we can see above, for 24 and 30 hpi the abortive_state is unknown. We would like to use biolord-classify to classify these cells.
By calling biolord.Biolord.setup_anndata() we set the semi-supervised attributes used for disentanglement.
The function takes as input:
adata: the adata object for the setting.ordered_attributes_keys: the keys inanndata.AnnData.obsoranndata.AnnData.obsmdefining ordered attributes.categorical_attributes_keys: the keys inanndata.AnnData.obsdefining categorical attributes.categorical_attributes_missing: a dict containing allcategorical_attributes_keysas keys. For each key, the value is the category that should be treated as the unknown label (if it contains missing labels) otherwise set to None. Here we will pass"abortive_state":"Unknown"and set the rest toNone.
biolord.Biolord.setup_anndata(
adata,
ordered_attributes_keys=["stress_score"],
categorical_attributes_keys=["time_int", "abortive_state", "zone"],
categorical_attributes_missing={"time_int": None, "abortive_state": "Unknown", "zone": None},
)
adata
AnnData object with n_obs × n_vars = 3024 × 1949
obs: 'barcode', 'mouse', 'marker', 'time', 'infected', 'experiment', 'coarse_time', 'MB', 'eta', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'mt_qc', 'SUMPBA', 'n_counts', 'n_genes', 'mus_rRNA', 'pba_rRNA', 'pba_rRNA_fraction', 'nCount_PBA', 'nFeature_PBA', 'nCount_MUS', 'nFeature_MUS', 'RNA_snn_res.0.2', 'seurat_clusters', 'MBinfected', 'cluster_names', 'ident', 'eta_normalized', 'nCounts_tot', 'normalized_PBA', 'coarse_time_orig', 'zone', 'status', 'status_control', 'time_int', 'pseudotime', 'abortive', 'abortive_state', 'stress_score', 'split_random', '_indices', '_scvi_time_int', '_scvi_abortive_state', '_scvi_zone'
var: 'org', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'abortive_state_colors', 'coarse_time_colors', 'hvg', 'log1p', 'neighbors', 'pca', 'status_colors', 'status_control_colors', 'umap', 'zone_colors', '_scvi_uuid', '_scvi_manager_uuid'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts', 'scaledata'
obsp: 'connectivities', 'distances'
Run biolord-classify#
Instantiate a Biolord model#
We instantiate the model given the module_params.
These are parameters required to construct the model’s module, the various networks included in a biolord-classify model.
To the parameters defined to train a classic biolord model we add values related to the classification task:
classification_penalty: penalty value for accuracy in training a regressor for ordered classes.classifier_penalty: the penalty weight of the classification term in the final loss objective.classify_all: a bool stating whether a classifier is trained for all attributes or only those with missing labels.classifier_dropout_rate: dropout rate for the classifiers
module_params = {
"decoder_width": 512,
"decoder_depth": 4,
"attribute_nn_width": 512,
"attribute_nn_depth": 4,
"use_batch_norm": False,
"use_layer_norm": False,
"unknown_attribute_noise_param": 1e-1,
"seed": 42,
"n_latent_attribute_ordered": 16,
"n_latent_attribute_categorical": 4,
"gene_likelihood": "normal",
"loss_regression": "normal",
"reconstruction_penalty": 1e1,
"unknown_attribute_penalty": 1e2,
"attribute_dropout_rate": 0.05,
"eval_r2_ordered": False,
"classifier_penalty": 1e1,
"classification_penalty": 0,
"classify_all": False,
"classifier_dropout_rate": 0.05,
}
To instantiate the BiolordClassifyModule, we pass train_classifiers=True to biolord.Biolord model.
model = biolord.Biolord(
adata=adata,
n_latent=32,
model_name="spatiotemporal_malaria_abortive",
train_classifiers=True,
module_params=module_params,
split_key="split_random",
)
Train the model#
To train the model we provide trainer_params. These are paramters which dictate the training regime, e.g., learning rate, weight decay and scheduler type.
trainer_params = {
"n_epochs_warmup": 0,
"latent_lr": 1e-3,
"latent_wd": 1e-4,
"decoder_lr": 1e-4,
"decoder_wd": 1e-4,
"attribute_nn_lr": 1e-2,
"attribute_nn_wd": 4e-8,
"step_size_lr": 90,
"cosine_scheduler": True,
"scheduler_final_lr": 1e-5,
}
model.train(
max_epochs=200,
batch_size=256,
plan_kwargs=trainer_params,
early_stopping=True,
early_stopping_patience=20,
check_val_every_n_epoch=10,
num_workers=1,
enable_checkpointing=False,
)
Epoch 90/200: 45%|████▌ | 90/200 [02:00<02:27, 1.34s/it, v_num=1, val_generative_mean_accuracy=0.963, val_generative_var_accuracy=0.802, val_biolord_metric=0.916, val_reconstruction_loss=0.728, val_unknown_attribute_penalty_loss=26.5, val_classification_accuracy=0.982, val_regression_r2_accuracy=0, val_regression_mse=0, val_classification_loss=0.395, generative_mean_accuracy=0, generative_var_accuracy=0, biolord_metric=0, reconstruction_loss=0.631, unknown_attribute_penalty_loss=31.4, classification_accuracy=0, regression_r2_accuracy=0, regression_mse=0, classification_loss=0.326]
Monitored metric val_biolord_metric did not improve in the last 20 records. Best score: 0.925. Signaling Trainer to stop.
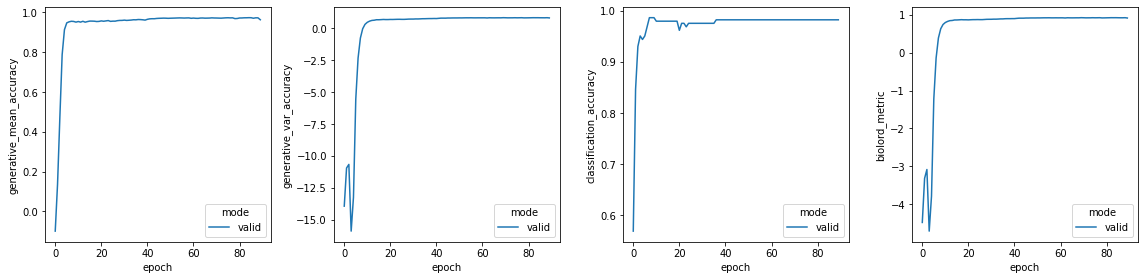
vals = ["generative_mean_accuracy", "generative_var_accuracy", "classification_accuracy", "biolord_metric"]
fig, axs = plt.subplots(nrows=1, ncols=len(vals), figsize=(4 * len(vals), 4))
model.epoch_history = pd.DataFrame.from_dict(model.training_plan.epoch_history)
for i, val in enumerate(vals):
sns.lineplot(
x="epoch",
y=val,
hue="mode",
data=model.epoch_history[model.epoch_history["mode"] == "valid"],
ax=axs[i],
)
plt.tight_layout()
Get the Abortive state classification#
Given the trained model we turn to classify cells at 24 and 30 hpi as Abortive/Productive.
idx_predict = np.where((adata.obs["coarse_time"] != "36 hpi"))[0]
adata_predict = adata[idx_predict].copy()
labels_color = {
"coarse_time": "coarse time",
"zone": "zone",
"abortive_state_classification": "abortive (classification)",
"abortive_state": "abortive (label)",
}
abortive_color = {
"Abortive": "#ff7c7b",
"Productive": "#7bbcff",
"Unknown": "#BDBABB",
}
coarse_time_colors = {"24 hpi": "#B99B6B", "30 hpi": "#F1DBBF", "36 hpi": "#AA5656"}
save_names = {"all": "all"}
datasets = {}
classification = {}
dataset = model.get_dataset(adata)
classification = model.module.classify(dataset["X"])
for attribute_, vals_ in classification.items():
if attribute_ in model.categorical_attributes_map:
inv_map = {v: k for k, v in model.categorical_attributes_map[attribute_].items()}
adata.obs[f"{attribute_}_classification"] = [inv_map[k] for k in vals_.argmax(dim=-1).cpu().numpy()]
else:
adata.obs[f"{attribute_}_classification"] = vals_[0].detach().cpu().numpy()
for attribute_, vals_ in classification.items():
adata.obs[f"{attribute_}_classification"] = adata.obs[f"{attribute_}_classification"].astype("category")
adata.obs[f"{attribute_}_classification_str"] = adata.obs[f"{attribute_}_classification"].copy()
adata.obs[f"{attribute_}_classification"] = adata.obs[f"{attribute_}_classification"].cat.rename_categories(
lambda x: str(x) + "\n(biolord-classify)"
)
sc.tl.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
fig, axs = plt.subplots(1, 3, figsize=(15, 4))
adata.uns["abortive_state_classification_colors"] = adata.uns["abortive_state_colors"][:2]
for i, c in enumerate(["coarse_time", "abortive_state", "abortive_state_classification"]):
sc.pl.umap(adata, color=[c], ax=axs[i], show=False)
axs[i].set_axis_off()
plt.tight_layout()
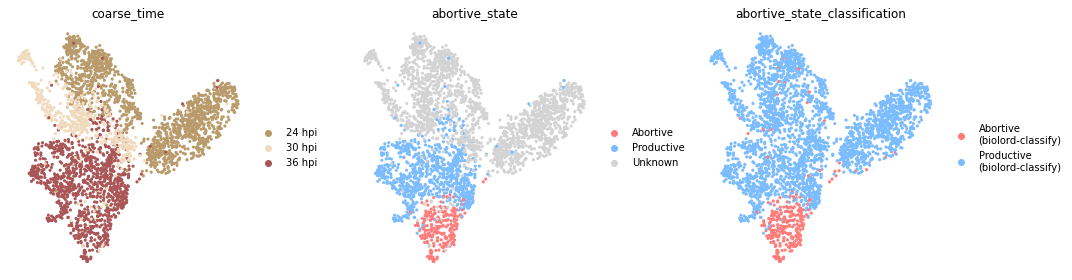
Evaluate classification#
We start by validating the classification accuracy with respect to labeled cells (36 hpi).
acc = (
adata[adata.obs["abortive_state"].isin(["Abortive", "Productive"])].obs["abortive_state"]
== adata[adata.obs["abortive_state"].isin(["Abortive", "Productive"])].obs["abortive_state_classification_str"]
).sum() / adata.obs["abortive_state"].isin(["Abortive", "Productive"]).sum()
print(f"abortive state classification accuracy is: {acc:.2f}")
abortive state classification accuracy is: 1.00
Population ratio#
Next we can look at the proportion of the Abortive/Productive populations.
Note that according to [AZLBH+22] at earlier time points the Abortive population is expected to be small, \(\sim2-5\%\).
hpis = {}
for hpi in adata.obs["coarse_time"].cat.categories:
hpis[hpi] = (
adata[adata.obs["coarse_time"].isin([hpi])].obs["abortive_state_classification"].value_counts()
/ adata[adata.obs["coarse_time"].isin([hpi])].shape[0]
).to_dict()
df_hpis = pd.DataFrame(hpis).T.reset_index()
df_hpis
| index | Productive\n(biolord-classify) | Abortive\n(biolord-classify) | |
|---|---|---|---|
| 0 | 24 hpi | 0.984160 | 0.015840 |
| 1 | 30 hpi | 0.942740 | 0.057260 |
| 2 | 36 hpi | 0.784857 | 0.215143 |
Spatial and parasite mRNA distribution#
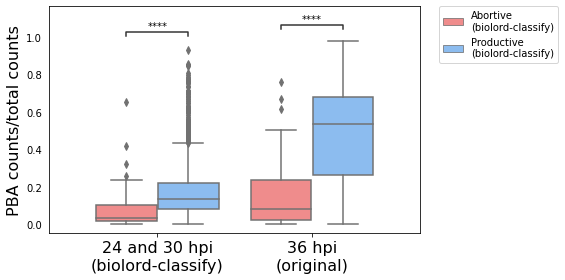
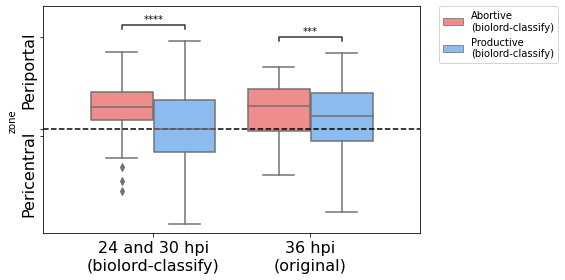
Following the properties of 36 hpi we expect the abortive population to be Periportally biased and present low parasite mRNA counts.
adata.obs["label_type"] = "biolord-classify"
adata.obs.loc[adata.obs["coarse_time"] == "36 hpi", "label_type"] = "original"
pairs = [
[("biolord-classify", "Abortive\n(biolord-classify)"), ("biolord-classify", "Productive\n(biolord-classify)")],
[("original", "Abortive\n(biolord-classify)"), ("original", "Productive\n(biolord-classify)")],
]
fig, ax = plt.subplots(1, 1, figsize=(8, 4))
df = adata.obs[
[
"label_type",
"abortive_state_classification",
"normalized_PBA",
"eta_normalized",
"zone",
"coarse_time",
"pseudotime",
]
]
plotting_parameters = {
"data": df,
"x": "label_type",
"y": "normalized_PBA",
"hue": "abortive_state_classification",
"palette": adata.uns["abortive_state_classification_colors"],
}
sns.boxplot(**plotting_parameters)
# Add annotations
annotator = Annotator(ax, pairs, **plotting_parameters)
annotator.configure(test="Mann-Whitney", comparisons_correction="Benjamini-Hochberg").apply_and_annotate()
ax.set_xlabel("")
ax.set_ylabel("PBA counts/total counts", fontsize=16)
ax.set_xticklabels(["24 and 30 hpi\n(biolord-classify)", "36 hpi\n(original)"], fontsize=16)
ax.yaxis.set_ticks_position("none")
plt.legend(bbox_to_anchor=(1.05, 1), loc="upper left", borderaxespad=0)
plt.tight_layout()
p-value annotation legend:
ns: p <= 1.00e+00
*: 1.00e-02 < p <= 5.00e-02
**: 1.00e-03 < p <= 1.00e-02
***: 1.00e-04 < p <= 1.00e-03
****: p <= 1.00e-04
original_Abortive
(biolord-classify) vs. original_Productive
(biolord-classify): Mann-Whitney-Wilcoxon test two-sided with Benjamini-Hochberg correction, P_val:2.170e-53 U_stat=3.396e+04
biolord-classify_Abortive
(biolord-classify) vs. biolord-classify_Productive
(biolord-classify): Mann-Whitney-Wilcoxon test two-sided with Benjamini-Hochberg correction, P_val:8.192e-11 U_stat=2.253e+04
fig, ax = plt.subplots(1, 1, figsize=(8, 4))
plotting_parameters = {
"data": df,
"x": "label_type",
"y": "eta_normalized",
"hue": "abortive_state_classification",
"palette": adata.uns["abortive_state_classification_colors"],
}
sns.boxplot(**plotting_parameters)
# Add annotations
annotator = Annotator(ax, pairs, **plotting_parameters)
annotator.configure(test="Mann-Whitney", comparisons_correction="Benjamini-Hochberg").apply_and_annotate()
th = adata[adata.obs["zone"] == "Pericentral"].obs["eta_normalized"].max()
ax.axhline(th, linestyle="--", color="black")
ax.set_xlabel("")
ax.set_ylabel("zone")
ax.set_yticks([0.48, th, 0.98])
ax.set_yticklabels(["Pericentral", "", "Periportal"], rotation=90, fontsize=16)
ax.set_xticklabels(["24 and 30 hpi\n(biolord-classify)", "36 hpi\n(original)"], fontsize=16)
plt.legend(bbox_to_anchor=(1.05, 1), loc="upper left", borderaxespad=0)
plt.tight_layout()
plt.show()
p-value annotation legend:
ns: p <= 1.00e+00
*: 1.00e-02 < p <= 5.00e-02
**: 1.00e-03 < p <= 1.00e-02
***: 1.00e-04 < p <= 1.00e-03
****: p <= 1.00e-04
original_Abortive
(biolord-classify) vs. original_Productive
(biolord-classify): Mann-Whitney-Wilcoxon test two-sided with Benjamini-Hochberg correction, P_val:3.056e-04 U_stat=1.143e+05
biolord-classify_Abortive
(biolord-classify) vs. biolord-classify_Productive
(biolord-classify): Mann-Whitney-Wilcoxon test two-sided with Benjamini-Hochberg correction, P_val:6.546e-05 U_stat=6.396e+04
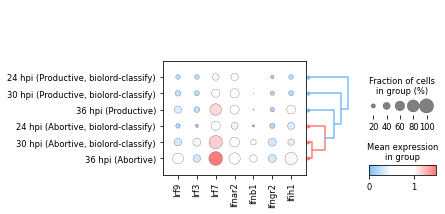
Interferon response#
At last, we can evaluate gene interferon response trajectory using representative genes.
adata.obs["time_abortive_classification"] = (
adata.obs["coarse_time"].astype(str) + " (" + adata.obs["abortive_state_classification"].astype(str) + ")"
)
adata.obs["time_abortive_classification"] = adata.obs["time_abortive_classification"].astype("category")
abortive_color = {
"Abortive": "#ff7c7b",
"Productive": "#7bbcff",
}
df_ifnr = adata.obs[["time_abortive_classification"]].reset_index()
for gene in ["Irf9", "Irf3", "Irf7", "Ifnar2", "Ifnb1", "Ifngr2", "Ifih1"]:
df_ifnr.loc[:, gene] = adata.X[:, adata.var_names.isin([gene])]
df_ifnr = df_ifnr.groupby("time_abortive_classification").mean()
sc.tl.dendrogram(adata, groupby="time_abortive_classification", linkage_method="centroid")
axs = sc.pl.dotplot(
adata,
["Irf9", "Irf3", "Irf7", "Ifnar2", "Ifnb1", "Ifngr2", "Ifih1"],
groupby="time_abortive_classification",
cmap=sns.blend_palette([abortive_color["Productive"], "1", "1", abortive_color["Abortive"]], as_cmap=True),
dendrogram=True,
show=False,
return_fig=True,
)
yticklabels = axs.get_axes()["mainplot_ax"].get_yticklabels()
yticklabels_new = []
node_colors = []
for ti, tick in enumerate(yticklabels):
if "36" in tick.get_text():
yticklabels_new.append(" ".join(tick.get_text().split("\n")[:1]) + ")")
else:
yticklabels_new.append(" ".join(tick.get_text().split("\n")[:1]) + ", biolord-classify)")
if "Abortive" in tick.get_text():
node_colors.append(abortive_color["Abortive"])
else:
node_colors.append(abortive_color["Productive"])
axs.get_axes()["mainplot_ax"].set_yticklabels(yticklabels_new)
for li, line in enumerate(axs.get_axes()["group_extra_ax"].get_lines()):
line.set_color(node_colors[-(li + 2)])
nodes = axs.get_axes()["group_extra_ax"].get_yticks()
for ni, node in enumerate(nodes):
axs.get_axes()["group_extra_ax"].plot(0, node, ".", color=node_colors[ni])